In chemical peptide synthesis, cyclizations require various procedures more or less complex. Peptides are first synthesized as linear form before being cyclized. Peptides can be cyclized through different approaches.
Disulfide-bridges containing peptides
This covalent link occurs between thiols of 2 cysteines. Disulfide-bridges are natural modifications that form highly structured peptides and are generally mandatory for the biological function of peptides. Disulfide-bridged peptides are widely present in Nature such as in animal venoms, plants…
The synthetic production of disulfide-bridged peptides is challenging especially when several disulfide-bridges must be formed. The synthesis usually starts with the production of the linear peptide which is then oxidized to allow cysteines connections. In the presence of several cysteines, peptides can naturally cyclize correctly and spontaneously but they can also form various conformers with a mixture of correctly and incorrectly folded peptides. The production of disulfide-bridged peptides is historically the core expertise of SB-PEPTIDE. The team has acquired a profound expertise in this field for many years while working on animal venom peptides and already produced thousands of peptides containing multiple bridges.
SB-PEPTIDE can typically produce:
1 disulfide-bridged peptide thanks to simple oxidation
1 disulfide bridge + 1 free cysteine: the free cysteine can be protected while forming the desired disulfide bridge then deprotected
Multiple disulfide-bridged peptides: a linear peptide can be oxidized randomly so several forms of the peptide can appear. Then the desired form has to be confirmed by another analysis. Alternatively, SB-PEPTIDE can fold the peptide by using cysteines orthogonally protected (Acm, StBu…). Thanks to this procedure, cysteines can be connected to the other cysteines as wanted. The company has a deep expertise in this field to produce complex peptide with high yield, correct folding and fast turnaround time.
Head-to-tail cyclization consists in forming an amide bond between the C-terminus carboxyl with the N-terminus amine. It has been demonstrated to improve the oral stability of certain therapeutic peptides without affecting their biological activity. Bonds between a carboxyl or amine containing side chain of an amino acid and the C-terminus or the N-terminus are also known as head-to-sidechain and sidechain-to-tail cyclization.
Peptides with Lactam bridges
Lactam bridge is a type of peptide cyclization which refers to the bond between the amine side chain of lysine (or a derivative) and a carboxyl side chain of a glutamate or an aspartate.
SB-PEPTIDE can employ various Lysine derivatives to play on lengths. The company can incorporate lysine derivatives with longer or shorter side-chain such as Dap (Diaminopimelic acid), Dab (Diaminobutyric acid) and Orn (Ornithine). For example, SB-PEPTIDE synthesized MiniAp-4 peptide, a blood-brain barrier shuttle peptide, which is a cyclic peptide with a lactam bridge between Dap and Asp. Lactam bridges allow to stabilize alpha helices and may replace a less-stable disulfide bond.
Thioether-bond containing peptides
Thioether macrocyclization, which is typically conducted by the reaction between a 2-chloroacetyl group and the thiol moiety in a cysteine sidechain, is a classical methodology for peptides prepared by standard chemical synthesis.
Chemically constrained peptide scaffold
Peptides can be cyclized using a chemical linker which binds to several amino acids within a peptide. This strategy allows to impose a particular shape to the peptide making them incredibly stable as well as highly potent. The usual strategy consists in using a chemical linker containing two or more methyl bromide groups which react with thiol group of cysteine residues. Various bromide linkers are available with several distances. This allows to play on the peptide structure. Constrained peptide may be a monocyclic peptide, a bicyclic peptide or a tricyclic peptide.
Constrained peptides are very useful to mimic secondary structures and to optimize the role of peptides. Indeed, constrained peptides allow to increase the affinity, the selectivity and the stability of peptides. SB-PEPTIDE has a deep expertise in the production of such constrained peptides and can obtain rapidly a large number of constrained peptides.
Stapled peptide
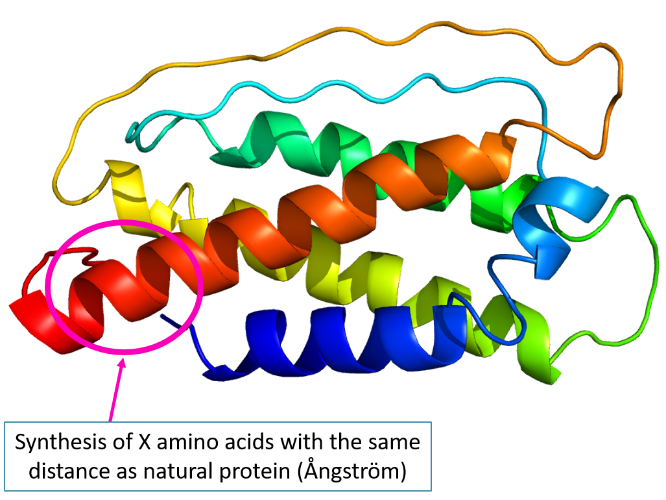
A stapled peptide has a constrained structure on its sidechain to induce or stabilize an alpha-helical conformation. This is obtained by forming a covalent link between sidechains of two close amino acids. Amino acids involved in the binding must have their sidechain outside of helical structure. The link can be obtain by introduction a chemical linker that will bind to each side chain, or by click-chemistry between 2 modified side-chains initially introduced. Stapled peptides are locked into a stabilized alpha helical structure. Different linker or clickable side chains may be used depending on the expected distance (precise Ångström, 1 turn, 2 turns…).
Stapled peptides are capable to mimic and to form stable alpha helical structures. These conformational structures are usually found at the interface of protein-protein interactions. Stapled peptide is notably used in research on therapeutic modulation of protein-protein interactions and to enhance the pharmacologic performance of peptides.
Cyclotides
Cyclotides are circular peptides with remarkable characteristics including a head-to-tail cyclized backbone and 6 cysteine residues forming cyclic-cystine-knot motif (CCK) by three disulfide bonds. Cyclotides show potent cytotoxic activity that’s why they represent novel range of cytotoxic agents.
SB-PEPTIDE is able to produce cyclotides such as Cycloviolacin O2.
Conformational structure reproduction of a peptide
Thanks to all cyclization methods, SB-PEPTIDE is able to reproduce conformational regions (conformational epitopes) of a protein and to modulate distances (Ångström, turn…). This can be highly valuable in the development of short protein-based vaccines.
Example:
- Interleukine 7:
Get in touch
SB-PEPTIDE has an extensive expertise in peptide folding as well as modification of folded peptides (fluorescent or biotin labeling for example). Contact us to discuss about the cyclic peptide you wish to produce.
1- Zoukimian C et al. Bioorg Med Chem. 27(1):247-253 (2019)
The scorpion toxin AmmTx3 is a specific blocker of Kv4 channels. It was shown to have interesting potential for neurological disorders. In this study, we report the first chemical synthesis of AmmTx3 by using the native chemical ligation strategy and validate its biological activity. We determined its 3D structure by nuclear magnetic resonance spectroscopy, and pointed out that AmmTx3 possesses the well-known CSαβ structural motif, which is found in a large number of scorpion toxins. Overall, this study establishes an easy synthetic access to biologically active AmmTx3 toxin.
2- Clairfeuille T et al. Science. 363(6433):eaav8573 (2019)
Fast inactivation of voltage-gated sodium (Nav) channels is essential for electrical signaling, but its mechanism remains poorly understood. Here we determined the structures of a eukaryotic Nav channel alone and in complex with a lethal α-scorpion toxin, AaH2, by electron microscopy, both at 3.5-angstrom resolution. AaH2 wedges into voltage-sensing domain IV (VSD4) to impede fast activation by trapping a deactivated state in which gating charge interactions bridge to the acidic intracellular carboxyl-terminal domain. In the absence of AaH2, the S4 helix of VSD4 undergoes a ~13-angstrom translation to unlatch the intracellular fast-inactivation gating machinery. Highlighting the polypharmacology of α-scorpion toxins, AaH2 also targets an unanticipated receptor site on VSD1 and a pore glycan adjacent to VSD4. Overall, this work provides key insights into fast inactivation, electromechanical coupling, and pathogenic mutations in Nav channels.
3- Tzakoniati F et al. Cell Chem Biol. 27(3):306-313.e4 (2020)
Voltage-gated sodium (Nav) channels respond to changes in the membrane potential of excitable cells through the concerted action of four voltage-sensor domains (VSDs). Subtype Nav1.7 plays an important role in the propagation of signals in pain-sensing neurons and is a target for the clinical development of novel analgesics. Certain inhibitory cystine knot (ICK) peptides produced by venomous animals potently modulate Nav1.7; however, the molecular mechanisms underlying their selective binding and activity remain elusive. This study reports on the design of a library of photoprobes based on the potent spider toxin Huwentoxin-IV and the determination of the toxin binding interface on VSD2 of Nav1.7 through a photocrosslinking and tandem mass spectrometry approach. Our Huwentoxin-IV probes selectively crosslink to extracellular loop S1-S2 and helix S3 of VSD2 in a chimeric channel system. Our results provide a strategy that will enable mapping of sites of interaction of other ICK peptides on Nav channels.
4- Nicolas S et al. Taxins (Basel). 11(6):367 (2019)
Phlotoxin-1 (PhlTx1) is a peptide previously identified in tarantula venom (Phlogius species) that belongs to the inhibitory cysteine-knot (ICK) toxin family. Like many ICK-based spider toxins, the synthesis of PhlTx1 appears particularly challenging, mostly for obtaining appropriate folding and concomitant suitable disulfide bridge formation. Herein, we describe a procedure for the chemical synthesis and the directed sequential disulfide bridge formation of PhlTx1 that allows for a straightforward production of this challenging peptide. We also performed extensive functional testing of PhlTx1 on 31 ion channel types and identified the voltage-gated sodium (Nav) channel Nav1.7 as the main target of this toxin. Moreover, we compared PhlTx1 activity to 10 other spider toxin activities on an automated patch-clamp system with Chinese Hamster Ovary (CHO) cells expressing human Nav1.7. Performing these analyses in reproducible conditions allowed for classification according to the potency of the best natural Nav1.7 peptide blockers. Finally, subsequent in vivo testing revealed that intrathecal injection of PhlTx1 reduces the response of mice to formalin in both the acute pain and inflammation phase without signs of neurotoxicity. PhlTx1 is thus an interesting toxin to investigate Nav1.7 involvement in cellular excitability and pain.
5- Goncalves T C et al. Br J Pharmacol. 176(9):1298-1314 (2019)
Background and purpose: The NaV 1.7 channel is highly expressed in dorsal root ganglia of the sensory nervous system and plays a central role in the pain signalling process. We investigated a library prepared from original venoms of 117 different animals to identify new selective inhibitors of this target.
Experimental approach: We used high throughput screening of a large venom collection using automated patch-clamp experiments on human voltage-gated sodium channel subtypes and then in vitro and in vivo electrophysiological experiments to characterize the active peptides that have been purified, sequenced, and chemically synthesized. Analgesic effects were evaluated in vivo in mice models.
Key results: We identified cyriotoxin-1a (CyrTx-1a), a novel peptide isolated from Cyriopagopus schioedtei spider venom, as a candidate for further characterization. This 33 amino acids toxin belongs to the inhibitor cystine knot structural family and inhibits hNaV 1.1-1.3 and 1.6-1.7 channels in the low nanomolar range, compared to the micromolar range for hNaV 1.4-1.5 and 1.8 channels. CyrTx-1a was 920 times more efficient at inhibiting tetrodotoxin (TTX)-sensitive than TTX-resistant sodium currents recorded from adult mouse dorsal root ganglia neurons and in vivo electrophysiological experiments showed that CyrTx-1a was approximately 170 times less efficient than huwentoxin-IV at altering mouse skeletal neuromuscular excitability properties. CyrTx-1a exhibited an analgesic effect in mice by increasing reaction time in the hot-plate assay.
Conclusions and implications: The pharmacological profile of CyrTx-1a paves the way for further molecular engineering aimed to optimize the potential antinociceptive properties of this peptide.
Peptidic toxins that target specifically mammalian channels and receptors can be found in the venom of animals. These toxins are rarely used directly as tools for biochemical experiments, and need to be modified via the attachment of chemical groups (e.g., radioactive or fluorescent moieties). Ideally, such modifications should maintain the toxin specificity and affinity for its target. With the goal of obtaining fluorescent derivatives of BeKm-1, a toxin from the scorpion species Buthus eupeus that selectively inhibits the voltage-gated potassium ion channel hERG, we produced four active analogues using a model of BeKm-1 docking to the outer mouth of the channel. In these BeKm-1 analogues, the natural peptide was linked to the fluorescent cyanine 5 (Cy5) probe via four different linkers at Arg1 or Arg/Lys27. All analogues retained their specificity towards the hERG channel in electrophysiological experiments but displayed a lesser affinity. These results validate our strategy for designing toxin analogues and demonstrate that different chemical groups can be attached to different residues of BeKm-1.
7- Montnach J et al. Br J Pharmacol. 178(13):2632-2650 (2021)
Background and purpose: Protoxin II (ProTx II) is a high affinity gating modifier that is thought to selectively block the Nav 1.7 voltage-dependent Na+ channel, a major therapeutic target for the control of pain. We aimed at producing ProTx II analogues entitled with novel functionalities for cell distribution studies and biochemical characterization of its Nav channel targets.
Experimental approach: We took advantage of the high affinity properties of the peptide, combined to its slow off rate, to design a number of new tagged analogues useful for imaging and biochemistry purposes. We used high-throughput automated patch-clamp to identify the analogues best matching the native properties of ProTx II and validated them on various Nav -expressing cells in pull-down and cell distribution studies.
Key results: Two of the produced ProTx II analogues, Biot-ProTx II and ATTO488-ProTx II, best emulate the pharmacological properties of unlabelled ProTx II, whereas other analogues remain high affinity blockers of Nav 1.7. The biotinylated version of ProTx II efficiently works for the pull-down of several Nav isoforms tested in a concentration-dependent manner, whereas the fluorescent ATTO488-ProTx II specifically labels the Nav 1.7 channel over other Nav isoforms tested in various experimental conditions.
Conclusions and implications: The properties of these ProTx II analogues as tools for Nav channel purification and cell distribution studies pave the way for a better understanding of ProTx II channel receptors in pain and their pathophysiological implications in sensory neuronal processing. The new fluorescent ProTx II should also be useful in the design of new drug screening strategies.
Cookies settings
This website uses cookies to ensure the operation and security of the website.
If you click on “Accept all”, we and third-party providers will process your personal data and store information (e.g. through cookies) on your device or access it. By clicking on the “Settings” button, you can view details of the processing, set your preferences, and reject processing operations that require consent.
Functional
Toujours activé
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Préférences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistiques
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.